The TikTok version of cortisol belly fat is wrong. Not about whether it exists, but about how it works. The actual mechanism is more specific, more surprising, and considerably harder to reverse than the wellness industry suggests.
Cortisol does not simply cause the body to produce more belly fat. Under chronic elevation, it preferentially relocates fat from peripheral deposits to visceral tissue through glucocorticoid receptor density differences in abdominal fat cells. It simultaneously drives cravings for calorie-dense foods through brain reward pathways, and disrupts leptin signaling so the satiety signal stops registering. The three mechanisms act together. Conventional caloric restriction often fails to address stress-related abdominal fat because it does not interrupt the cortisol pattern driving the redistribution.
There is a Johns Hopkins endocrinologist named Rexford Ahima who said something that deserved more attention than it got: the idea that high cortisol straightforwardly causes belly fat does not have strong evidence behind it. He was right. Not because the cortisol-abdomen connection is a myth, but because the mechanism is more precise than the popular narrative captures. Cortisol does not broadcast fat gain broadly. It targets a specific tissue, through a specific receptor, in a specific location. Understanding that precision changes what you actually do about it.
What follows is not a defense of the wellness industry's cortisol narrative. It is an attempt to explain what the research actually shows, which is simultaneously more modest in some ways and more specific in others. The redistribution mechanism is real. The craving mechanism is well-documented. The satiety disruption is measurable. And there is a feedback loop inside visceral fat itself that almost no popular account mentions. That loop is why the fat stays.
Part 1: The Redistribution Mechanism
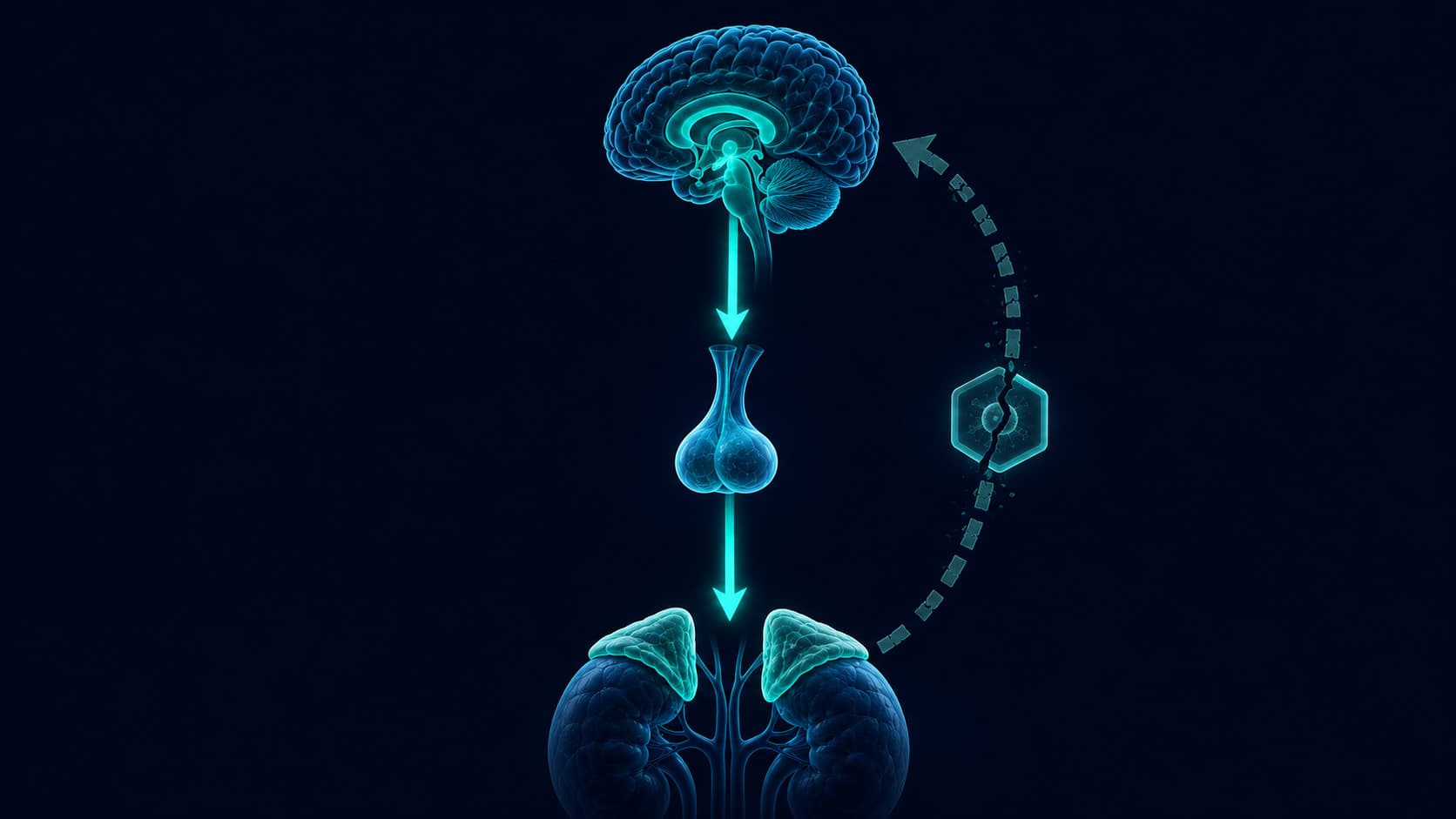
Fat cells are not uniform. Their glucocorticoid receptor density varies significantly by location in the body. Visceral adipose tissue, the fat that surrounds the internal organs in the abdominal cavity, has substantially higher receptor density than subcutaneous fat, which sits just beneath the skin at the hips, thighs, and arms. When cortisol circulates, it binds to these receptors and activates lipoprotein lipase, the enzyme that pulls triglycerides out of the bloodstream and into fat cells.
Because visceral fat has more receptors, it receives a stronger signal. Fat that might otherwise be stored peripherally gets routed centrally. This is not necessarily a net increase in total body fat. Under certain conditions of chronic stress, the scale may not change significantly while the distribution shifts markedly toward the abdomen. The body is not gaining more fat in every case. It is moving existing fat to a more metabolically dangerous location.
Visceral fat carries greater metabolic risk than subcutaneous fat because of its proximity to the portal circulation supplying the liver. Free fatty acids released by visceral fat cells go directly to the liver, contributing to insulin resistance and dyslipidemia at rates that peripheral fat does not. The location matters not just aesthetically. It matters metabolically.
Björntorp reviewed the relationship between chronic HPA axis activation and visceral adiposity across multiple human populations. Individuals with flattened diurnal cortisol rhythms and elevated afternoon cortisol consistently showed disproportionate visceral fat accumulation. The review proposed that HPA dysregulation, not caloric excess in isolation, drives the metabolic syndrome phenotype observed in chronic stress populations. The mechanism runs through glucocorticoid receptor-mediated activation of visceral adipose tissue specifically.
“Visceral fat accumulation appears to be a consequence of dysregulated HPA axis activity, with elevated and poorly rhythmic glucocorticoid exposure selectively activating lipid uptake in intra-abdominal adipose tissue through receptor-mediated mechanisms.”
Part 2: How Cortisol Rewrites What Your Brain Wants to Eat
Comfort food is not a personality trait. It is a pharmacological response that the brain initiates for a specific purpose. Corticotropin-releasing factor, the brain signal that starts the cortisol cascade, acts directly on the mesolimbic dopamine system. Under chronic stress, that circuit becomes sensitized to calorie-dense, high-fat, high-sugar foods in a way it is not sensitized to other food types.
The UCSF research group led by Mary Dallman examined in 2003 whether the craving shift under chronic stress had a functional relationship to HPA axis activity, specifically asking whether satisfying those cravings does something measurable to the stress signal itself.
Dallman and colleagues demonstrated that chronic stress activates a CRF-driven pathway that specifically increases consumption of calorie-dense foods, and that this consumption measurably suppresses HPA axis activity. The research reframed comfort eating not as a willpower failure but as a neurobiologically coherent self-regulation mechanism: the body uses calorie-dense food to temporarily reduce its own cortisol signal. The problem is that the foods most effective at doing this are also the most efficiently deposited in visceral fat.
“Ingestion of comfort food suppresses activity in the HPA axis, suggesting that chronic stress engages a neurobiological drive toward calorie-dense foods as a self-regulatory response to sustained glucocorticoid activity.”
The brain learns this association quickly. The more reliably a high-fat or high-sugar food reduces the stress signal, the more reliably stress will produce the craving for that food. The drive is not random appetite. It is a targeted biological strategy for temporary stress relief.
The irony of the mechanism is that the foods it reaches for are also the ones most efficiently converted to triglycerides and routed to the visceral deposits that Part 1 described. The craving system and the storage routing system are not parallel processes running independently. They are coordinated. Part 1 primes the storage. Part 2 delivers the material.

Part 3: Why the Fullness Signal Stops Working
Leptin tells the brain the body has enough stored energy. As fat mass increases, leptin production rises, the hypothalamus receives the signal, and appetite should fall. In a healthy system, this is the self-correcting mechanism that prevents runaway fat accumulation. Chronic cortisol elevation dismantles it at two points.
First, glucocorticoids reduce leptin receptor sensitivity in the hypothalamus, meaning the signal exists but the receiver is no longer responding. Second, the pro-inflammatory cytokines that elevated cortisol promotes, including IL-1 beta, further impair leptin signaling through separate pathways. The result is functional leptin resistance: fat stores are increasing, leptin is rising appropriately in response, but the brain is not reducing appetite because it cannot hear the message. The regulator is offline.
Sleep fragmentation, which chronically elevated evening cortisol produces, simultaneously raises ghrelin, the hunger-promoting hormone that works in opposition to leptin. The person arrives at the next morning already in a hunger surplus before they have eaten anything. Parts two and three do not take turns. They run simultaneously.
Adam and Epel reviewed the intersection of the stress response, mesolimbic reward pathways, and eating behavior. They documented that chronic stress drives two simultaneous processes: heightened motivation to consume calorie-dense foods through dopamine system sensitization, and reduced sensitivity to satiety signals through glucocorticoid interference with leptin and insulin signaling. The combination produces a pattern of elevated intake with low satiety response and preferential central fat storage.
“Glucocorticoids promote central fat deposition through peripheral receptor mechanisms while simultaneously sensitizing reward circuits to calorie-dense food cues and attenuating hypothalamic satiety signals, creating a convergent drive toward stress-induced central adiposity.”
The Feedback Loop Inside the Fat Itself
Almost no account of cortisol and belly fat explains what happens next, after the visceral fat has accumulated. Visceral fat cells express an enzyme called 11 beta-hydroxysteroid dehydrogenase type 1, commonly written as 11β-HSD1. This enzyme converts inactive cortisone into active cortisol locally, inside the fat tissue itself. The adipose tissue begins generating its own cortisol signal independent of the adrenal glands.
The more visceral fat accumulates, the more 11β-HSD1 is expressed, the more local cortisol is produced, and the more the glucocorticoid receptor-mediated storage mechanism in that same tissue is activated. This is a self-sustaining loop with no requirement for external stressors to maintain it. A person whose original stress has resolved may find that the cortisol signal driving continued visceral fat accumulation is now coming from the fat itself.
This is the mechanistic reason visceral fat is more resistant than subcutaneous fat. Subcutaneous fat does not express 11β-HSD1 at comparable levels. It is not generating its own cortisol. It responds to a systemic hormonal environment. Visceral fat creates that environment locally, which is why interventions that do not specifically address this tissue can produce disappointing results.
Visceral fat contains 11β-HSD1, an enzyme that converts inactive cortisone into active cortisol inside the fat tissue itself. This means belly fat is not just a product of stress. After accumulating, it actively generates the hormone responsible for keeping it there, independent of any external stressor.
Why Caloric Restriction Under Stress Often Makes This Worse
The natural response to weight gain is to eat less. For ordinary weight gain, this works with predictable proportionality. For stress-driven visceral fat accumulation, a 2010 UCLA study by Tomiyama and colleagues examined something the conventional diet narrative had not directly tested: whether caloric restriction itself affects the hormonal environment driving the problem. Their participants were randomized to different restriction levels, with salivary cortisol monitored throughout.
Tomiyama and colleagues randomized participants to caloric restriction conditions and monitored salivary cortisol over time. The group experiencing the most severe caloric restriction showed the largest cortisol increases, despite achieving weight loss. The researchers noted that restrictive dieting may inadvertently activate HPA axis responses that counteract the intended metabolic effect, particularly when pre-existing psychological stress is present. They suggested that cortisol regulation may need to precede or accompany dietary intervention for the approach to be effective.
“Caloric restriction was associated with increased cortisol output, with the most severe restriction producing the largest cortisol increases, suggesting that restrictive dieting may inadvertently activate stress responses that counteract weight loss efforts.”
The mechanism is evolutionarily coherent. Caloric restriction signals food scarcity. The HPA axis treats food scarcity as a stressor. It responds by elevating cortisol, which in a chronic state activates the same visceral fat redistribution mechanism the person is trying to reverse. Aggressive dieting under conditions of active psychological stress can amplify the exact hormonal pattern driving the problem. The fat responds not to the caloric math but to the cortisol signal.
Ordinary Weight Gain
- Fat distributed across the body
- Responds proportionally to caloric reduction
- Driven primarily by energy surplus
- Leptin signaling generally intact
- No self-sustaining hormonal feedback loop
Cortisol-Driven Visceral Fat
- Fat relocated preferentially to the abdomen
- Partially resistant to caloric restriction alone
- Driven by glucocorticoid receptor routing
- Leptin resistance active: satiety signal disrupted
- Visceral fat generates its own cortisol via 11β-HSD1
What the Research Points Toward Instead
The interventions with the most consistent support in the literature address the cortisol pattern rather than the caloric balance. The dietary component matters, but its effectiveness depends on the hormonal environment in which it operates.
- Sleep continuity before 1 AM: slow-wave sleep is the primary recovery window for the HPA axis and for glucocorticoid receptor resensitization. Sleep disruption maintains the hormonal state that drives visceral redistribution. No dietary intervention compensates for this.
- Morning light before screen exposure: circadian anchoring steepens the morning cortisol peak and lowers the afternoon baseline within days. A stronger diurnal rhythm reduces the chronic elevation that drives visceral routing.
- Protein-forward meals rather than aggressive restriction: protein supports stable blood glucose without triggering the scarcity signal that Tomiyama's research identified. It also has the highest satiety-per-calorie ratio, partially compensating for blunted leptin signaling.
- Moderate aerobic exercise, sustained over weeks: moderate intensity improves glucocorticoid receptor sensitivity over time. Chronic high-intensity training without adequate recovery can sustain HPA activation rather than reduce it.
- Addressing the cortisol rhythm before aggressive caloric restriction: Tomiyama's finding suggests sequencing matters. Attempting severe restriction under active psychological stress may not produce expected results and can amplify the problem.

The Cortisol Series Continues
The next article in the series examines what chronic cortisol elevation does to the brain: memory, focus, decision-making, and the mechanism behind stress-related cognitive fog.
Subscribe for the full seriesGetClariSync Body Desk
Editorial Research · Sports & Movement Science
The GetClariSync Body Desk reviews research in exercise physiology, recovery science, and sports nutrition. We follow journals including Medicine & Science in Sports & Exercise, the Journal of Applied Physiology, the British Journal of Sports Medicine, and the European Journal of Applied Physiology. We separate findings from trained-athlete populations from those relevant to recreational readers, and we flag when transferring a protocol across populations is unsupported. We are editorial researchers, not certified trainers, physiotherapists, or sports physicians — please consult a qualified professional before starting new exercise programs, especially with existing injuries, pregnancy, cardiovascular conditions, or chronic disease.